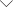近日,浙江大学物理学院第一性原理计算团队在计算物理与人工智能交叉领域取得突破。 该团队提出并实现了一种名为 DeepDM 的变分机器学习模型,用于直接求解电子结构问题中的基态密度矩阵,从而高效、高精度地获取材料与分子的物理性质。 这项开创性研究以《Variational Machine Learning Model for Electronic Structure Optimization via the Density Matrix》为题,于近期发表在国际顶级物理期刊《物理评论快报》(Phys. Rev. Lett. 135, 256403 (2025))上,为利用人工智能加速大规模量子力学计算开辟了全新路径。
在材料科学、化学和物理研究中,密度泛函理论(DFT)是计算电子结构、预测材料性质的核心工具。 然而,传统的 DFT 计算依赖于“自洽场”迭代方法,其核心步骤——哈密顿矩阵对角化——计算代价高昂,成为制约大规模、高通量模拟的瓶颈。 近年来,尽管机器学习已成功应用于预测 DFT 计算结果,但这些“数据驱动”模型严重依赖预先构建的、海量且成本高昂的计算数据集,且难以保证严格的物理约束。
从“拟合数据”到“求解方程”
该研究颠覆了传统思路。 他们提出的 DeepDM 模型不再学习已有的计算结果,而是直接学习如何“求解” Kohn-Sham 方程。 该模型的核心创新在于,将等变图神经网络与量子力学的变分原理深度融合,构建了一个端到端的物理信息机器学习框架。
具体而言,模型以原子结构(视为图网络的节点与边)为输入,通过精心设计的 等变图注意力网络,直接输出满足所有关键物理约束(厄米性、粒子数守恒、幂等性)的密度矩阵。 随后,利用 DFT 能量泛函作为唯一训练目标,通过反向传播直接优化神经网络参数,从而“无监督”地寻找到系统的基态能量与电子密度。 这一过程完全绕过了传统的哈密顿矩阵对角化步骤,也无需任何预先计算的数据集,实现了从“数据驱动”到“物理驱动”的根本性范式转变。
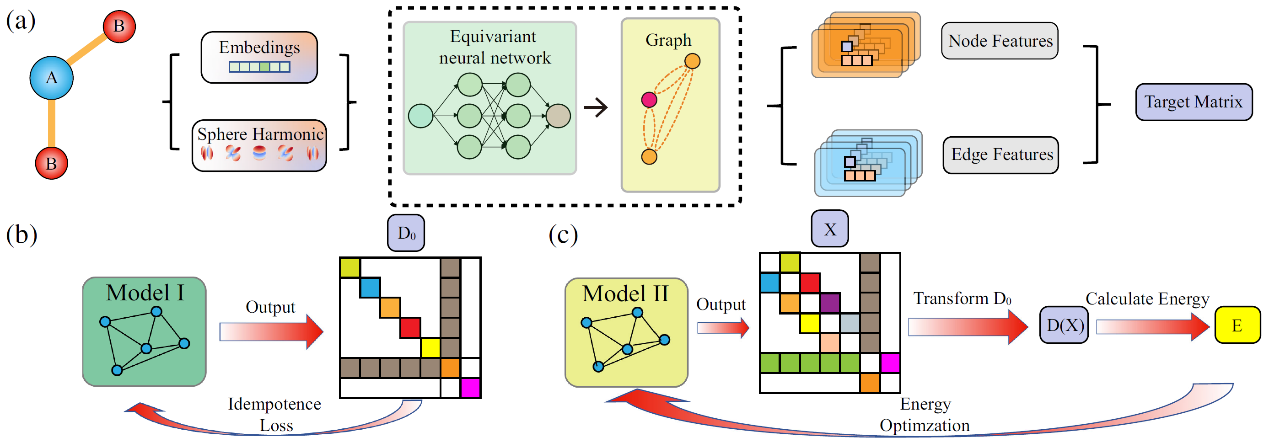
图1 DeepDM的主要工作流程
在神经网络中严格保持物理约束
确保神经网络输出的密度矩阵满足严格的物理约束是最大挑战之一。 研究团队创造性引入了指数参数化方法:首先训练一个网络生成满足幂等性等约束的初始密度矩阵;随后,由第二个网络预测一个反对称的修正矩阵,通过指数变换在保持所有物理约束不变的前提下,对初始矩阵进行优化微调,最终收敛到能量最低的基态密度矩阵。 这一精巧设计确保了整个优化过程始终在物理合理的参数空间内进行。
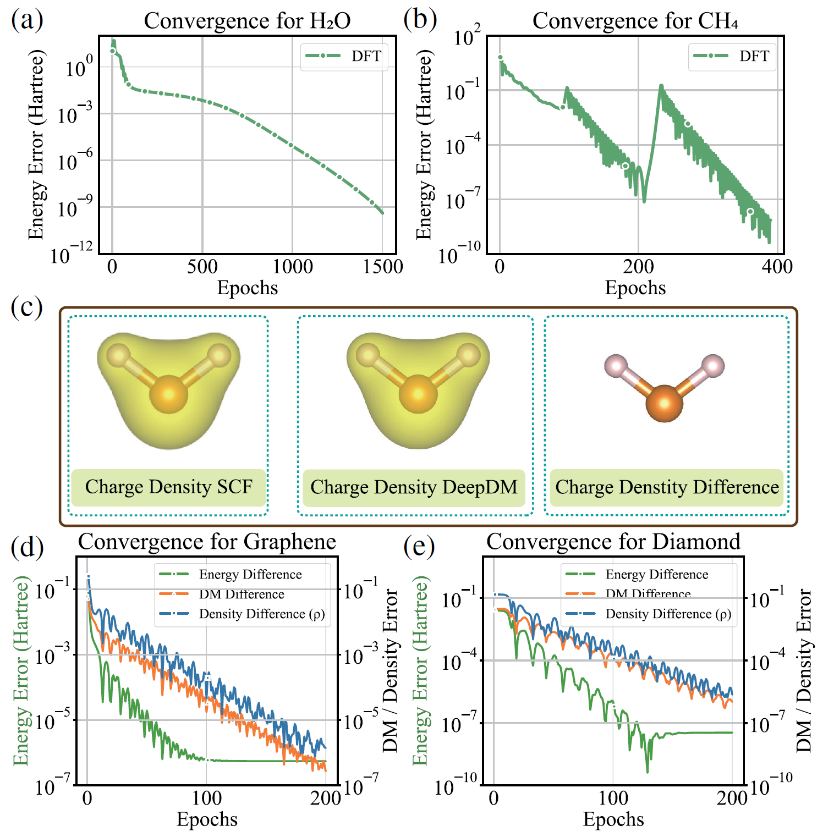
图2 DeepDM的验证
精度媲美标准方法,兼具泛化与扩展能力
研究团队在多个体系上验证了 DeepDM 的卓越性能。 对于水分子(H₂O)、甲烷(CH₄)等典型分子,DeepDM 优化得到的基态能量与电荷密度分布,与成熟的传统自洽场(SCF)计算结果高度吻合,误差可忽略不计。 更重要的是,该模型成功推广至石墨烯、金刚石等周期性扩展体系,同样展现出极高的计算精度。
图3 DeepDM的泛化测试
除了高精度,DeepDM 还表现出优秀的泛化与规模扩展能力。 研究显示,仅用原胞结构训练得到的模型,无需重新训练,即可准确预测更大超胞(如2×2×1石墨烯超胞)的电子结构性质,其预测精度远优于基于核心哈密顿量对角化的传统初始猜测方法。 这表明,DeepDM 不仅是一个高精度求解器,还能作为一个高效的、近乎收敛的“高级初始猜测”生成器,有望大幅减少后续传统 SCF 计算的迭代次数,从而显著降低大规模电子结构模拟的总计算成本。
该研究成果标志着人工智能与第一性原理计算融合迈入了新阶段。 DeepDM 模型首次展示了将第一性原理计算本身作为训练目标,从而实现“无数据”的高精度机器学习模拟的可行性。 这为解决科学计算中高质量数据稀缺的难题提供了全新思路。
相关成果以“《Variational Machine Learning Model for Electronic Structure Optimization via the Density Matrix》”为题发表于《物理评论快报》(Phys. Rev. Lett. 135, 256403 (2025))。论文第一作者为浙江大学物理学院博士生董鲁奇,通讯作者为陆赟豪教授,东方理工魏苏淮教授参与讨论,其他作者还包括之江实验室杨树祥研究员。该研究得到了国家重点研发计划等项目的支持。